
在研、上市激酶抑制剂的激酶组选择性
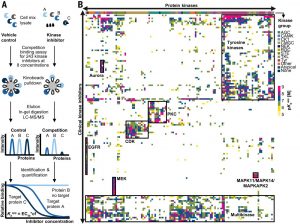
【新闻事件】:今天的《科学》杂志发表一篇文章系统测量现在已经上市的37个、和正在临床开发的250个激酶抑制剂的激酶组选择性。作者把四种肿瘤细胞打碎后用不同浓度的激酶抑制剂处理,然后用连在固相树脂的星形孢菌素把尚未被激酶抑制剂饱和的激酶吸附,这样可以计算每个激酶抑制剂与每种激酶的结合力。结果除了极少数药物如Tykerb,多数已经进入临床和上市药物都选择性欠佳,包括号称高选择性的激酶抑制剂。如dabrafenib号称选择性BRAF抑制剂,但和30个激酶有强于1微摩尔的结合力。作者利用这个激酶选择性网络数据为一些新靶点找到临床药物作为优质先导物,如有21个临床化合物击中抗炎新靶点SIK2。也为部分有脱靶活性的药物找到新的适应症,如golvatinib因为与FLT3结合所以可能用于治疗AML。
【药源解析】:激酶是个大家族,有518个成员。绝大多数激酶抑制剂都是与ATP结合腔结合,而激酶的这个结合腔大同小异,所以激酶组选择性是激酶抑制剂开发的一个重要技术障碍。现在已有多种测量激酶组(518个激酶中的绝大多数)选择性的技术如Kinative,而几乎每个激酶药物进入临床前都要测量激酶组选择性。但今天这个工作是第一次把所有在研、上市药物用同一个方法测量,所以更具可比性。至于为新靶点找已知配体、为药物找新适应症,其它测量选择性的技术都可以做到,Kinobead并无特别之处。
星形孢菌素是个选择性极差的激酶抑制剂。因为几乎所有激酶都与星形孢菌素高强度结合,所以除非某个激酶提前被选择性抑制剂饱和都会被星形孢菌素树脂捞出。然后把这些激酶用自由的星形孢菌素从树脂上洗脱下来、蛋白酶降解后用质谱测量每个蛋白特有多肽片段丰度,即可估算一个化合物对每个激酶的结合力。这个工作的一个有趣发现是二型抑制剂的选择性并没有比一型抑制剂好多少,这与现有观念有点差异,但三型抑制剂确实选择更好。这个工作用的是打碎细胞,所以一些在完整细胞中与激酶结合的结构蛋白可能已经分离。另外很多激酶需要磷酸化激活,打碎的细胞这些信号可能不存在。所以这个技术测出的选择性与在完整细胞的选择性不一定完全一致。虽然用完整细胞更科学,但这要求抑制剂/蛋白复合物的解离速度要比较慢才能可靠地比较活性,但不同化合物与不同蛋白的解离速度可能差很多。
从这个研究的结果看现在即使已经进入临床的激酶抑制剂选择性也强差人意(见图)。不仅多数药物有脱靶激酶活性,还与一些非激酶有交叉活性。象今年上市的所谓FLT3抑制剂midostaurin,整个一个激酶抑制剂中的花花公子,几乎与所有激酶有染。其AML疗效有多少与抑制FLT3有关只有上帝知道。对于用激酶抑制剂当作科研工具的基础研究科学家来说这种低选择性的危害要大得多。如果你的BRAF工具化合物抑制EGFR的能力比BRAF还强,你怎么用它研究BRAF功能?但是现在文献中有大量这类研究,所以要看到有人报道激酶A抑制剂能延年益寿,你首先要看看这个化合物选择性怎么样,多数情况也抑制B、C、D,所以与抑制A并无关系。
当然作为药物一旦证明疗效大于副作用,没人关心你有多少脱靶活性。这类似中国足球职业联赛刚开始的体能测试,当时有几个国家队主力都未能通过12分钟3200米的低标准。选择性差并不一定就不能成药,如同体能差不一定进不了国家队。但是选择性差令药理学数据与基因学证据脱钩,使开发不确定性增加、风险加大。从这个研究结果看现在激酶靶点确证和开发过程基本是一本糊涂账,能有37个激酶抑制剂上市要谢天、谢地、谢人。
美中药源原创文章,转载注明出处并添加超链接,商业用途需经书面授权。★更多深度解析访问《美中药源》~
★ 请关注《美中药源》微信公众号 ★

One Response to 在研、上市激酶抑制剂的激酶组选择性
发表评论
要发表评论,您必须先登录。

:何为生物类似药?")


:生物类似药 VS. 化学仿制药")



- 路人丙: 新药发现的低悬果实
- Pipi_WHU: 新药发现的低悬果实
- 路人丙: Galapagos放弃IPF资产、吉利德空手而归
- Tan, Chengfang: Galapagos放弃IPF资产、吉利德空手而归
- 配体效率遭到质疑 | 美中药源: 你笑我无知类药性本不存在?
- Songdanqing: 你笑我无知类药性本不存在?
- 路人丙: 中国新药的新时代
- 康, 荣明: 中国新药的新时代
合作伙伴









 微信号:美中药源
微信号:美中药源
Pingback: 激酶抑制剂药物路在何方? | 臧敬五博客