
从基因的角度解析心血管疾病风险(三)没人能逃避的动脉粥样硬化
By 沈骊 GB HealthWatch

第三章:没人能逃避的动脉粥样硬化
我们总觉得动脉粥样硬化是老年人的毛病,与自己无关,其实这一过程从孩童时期就开始了。对那些携带有家族性高胆固醇血症基因突变的人,胆固醇浓度从小就会很高,由于血管壁长期暴露在高胆固醇环境,这些人在二三十岁的年龄就可以有明显的动脉粥样硬化,四五十岁时发生心肌梗塞事件的几率会非常高。表面上看,血液中胆固醇浓度越高,出现动脉粥样硬化的时间越早,似乎高胆固醇就是问题的源头。然而大家是否注意到,其实动脉血管和静脉血管都暴露在相同浓度的胆固醇下,就连抽血检测胆固醇抽的也是静脉血。为什么我们没听说过静脉粥样硬化呢?事实上,如果将一段静脉血管移植到动脉血管的位置,比如心脏搭桥手术中将身体中一段静脉血管移植到冠状动脉位置,这一段移植来的静脉血管经一段时间后也会出现粥样硬化斑块。可见光光是胆固醇并不足以造成动脉粥样硬化,那么动脉粥样硬化的起始原因是什么呢?

图 1. 严重粥样硬化的动脉血管剖面图 。胆固醇和油脂沉积在动脉血管内皮细胞与平滑肌之间的夹层中。
动脉粥样硬化的形成过程
科学界并不完全知道动脉粥样硬化的起因和病理过程。一个被广泛接受的假说把动脉粥样硬化的过程描述成四步:第一步,动脉血管壁内膜的单层内皮细胞层发生了物理损伤,这是起始点。第二步,富含胆固醇的脂蛋白微粒从损伤处浸润进入血管壁内皮细胞层与平滑肌层之间的夹层,胆固醇滞留在夹层中,形成显微镜下可观察到的脂肪纹。第三步,沉积在夹层中的胆固醇被自由基氧化,触发免疫细胞前来清扫;免疫巨噬细胞由于吞咽了大量胆固醇,在显微镜下看起来像泡沫一样,因此称为泡沫样细胞;随着巨噬细胞死亡,其中的胆固醇和油脂泄漏出来聚合在一起,成为动脉粥样硬化中的 “粥” 的部分。第四步,血管壁中层的平滑肌细胞受到生物信号刺激向上迁移增生,把油脂和泡沫样细胞復盖在下面,这些平滑肌细胞又进一步纤维化和钙化,结果在 “粥”的上面形成一层磷酸钙的硬壳; 磷酸钙就是骨头的成分所以很硬,因此这种发生在动脉血管壁上的病变叫做动脉粥样硬化。
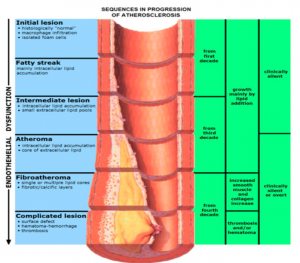
图2. 动脉粥样硬化的进程图。
在生命的前二十年,胆固醇和油脂就开始在血管壁夹层中逐渐积聚,这个阶段只有在显微镜下可以看到有脂肪纹,没有临床症状,自己不会有感觉,医生也无法诊断。从三十岁开始,一小部份人的血管开始出现稍微的狭窄,也没有症状。从四十岁以后,血管壁夹层中的堆聚物会变得越来越明显,相当一部分人的血管开始出现狭窄,极少一小部分人可能已经严重狭窄。超过五十岁以后,随着年龄的增长,越来越多的人的血管出现有狭窄区,也就有越来越多的人发生缺血性心脑血管病事件。这里我们可以看到,动脉粥样硬化是一个日积月累的过程,在一部份人身上这个过程发展的快些而另一部份人慢些。那些因素会加快动脉粥样硬化的进程呢?
血压的影响
为什么动脉粥样硬化只出现在动脉不出现在静脉?动脉血管中血压高是一个重要原因。动脉血管由三层结构组成,外壁由弹性纤维细胞组成,中间是平滑肌层,内层是由单层上皮细胞组成的内膜。粥样硬化通常只出现在弹性动脉和肌肉动脉上,而不会出现在小动脉上。主动脉,冠状动脉,颈动脉和股动脉是最早出现粥样硬化的部位。弹性动脉和肌肉动脉的共同特征是都有很厚的中间层平滑肌,用于控制血管的收缩和舒张从而改变血压。血压对血液循环很重要,没有血压血液就不能流动,就无法把含氧和营养物的血液挤压进入需要供氧的器官和组织,比如心肌和大脑等。静脉血管没有平滑肌中间层,不能收缩;当血液量增加时静脉血管壁会自动扩张变粗但不会增加血压。动脉血管壁的内膜所受到的血液压力几乎是静脉血管壁的10倍。动脉血管必须时时刻刻维持一定的压力来保障血液循环。血压升高可能加大对血管壁内膜的损伤。确实那些参与血管壁平滑肌舒张和控制血压的基因比如NOS3, GUCY1A1等,这些基因上的某些变异会显著增加心血管病风险。
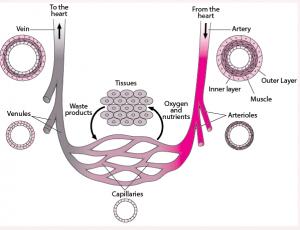
图3. 动脉和静脉血管的结构
血流动力学的影响
动脉血管中除了血压高以外,受血液流动冲击最大的位置也最容易出现粥样硬化斑块位置。观察基因改造后患上高胆固醇血症的动物模型发现,粥样硬化斑块并不是随机出现在动脉血管的任何区域,而是有非常固定的位置,总是在动脉血管急转弯处或分岔处,并且总是在弧形突出来的一侧。这些位置共同的血流动力学特征是层流减少剪应力低(low sheer stress),同时振荡流和湍流增加壁面拉应力大(high mural tensile stress)。相反在比较直的血管区段,血流主要是层流,剪应力高,血管壁内皮细胞顺着血流方向整齐紧密排列,血流中的脂蛋白微粒很难粘附到血管壁上,所以很难发生粥样硬化。而在血管转弯和分岔处,血管壁内皮细胞层受到来自湍流的不同方向的冲击和拉扯,受力不均而且壁面拉应力因此对血管壁的损伤较大。一旦有破损出现,血流中的脂蛋微粒就可能粘附到破损处并穿过内皮细胞层进入血管壁夹层。同时这样的流体动力学特征也会造成LDL微粒这样大小密度的颗粒更容易聚集在有湍流的区域,增加随机碰撞到血管壁损伤处的几率。因为血液流动分分秒秒都在进行中,易受损位置分分秒秒受着血流冲击,日积月累就会首先出现粥样硬化病变。
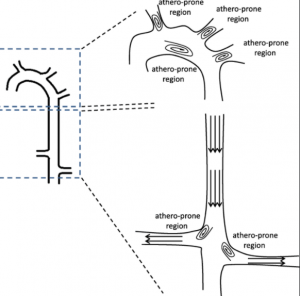
图4. 血液流动的力学特征决定了动脉粥样硬化出现的位置.
除了血压和血流强度会对血管壁内膜造成损伤外,吸烟和高血糖也会增加血管壁内膜的易损性。吸烟会增加血液中氧化自由基的浓度,可以直接损伤血管壁内膜细胞。
高胆固醇的影响
有了动脉血管壁损伤在先,血液中的胆固醇高低就很大程度决定了动脉粥样硬化的快慢。我们可以把血液中的脂蛋白微粒想像成运载着油脂和胆固醇的小球。 在第二章中我们讲过血液中的脂蛋白微粒分成五种,其中低密度脂蛋白微粒LDL是载有胆固醇最多的一种,每一个LDL微粒运载大约3000个胆固醇分子。由于血管转弯和分岔处的动力学特征,个头较小密度较高的LDL微粒可能更多滞留在转弯处的湍流里,相对于其他个头更大的脂蛋白微粒,LDL粘附到血管壁的破损处的几率会更高。血液中大部分胆固醇存在于LDL微粒上,体检报告上的低密度胆固醇LDL-C指的就是被LDL微粒运载着的胆固醇。不难想象LDL-C升高的结果会使更多的胆固醇随机粘附到血管壁的破损处,如果没有被及时清除,最终会在那些有破损的位置形成粥样硬化斑块。另外,比LDL微粒大一点的残体微粒,包括IDL和乳糜微粒残体,所含胆固醇量也很高,也相对容易滞留在血管转弯和分岔的位置,只是通常情况下残体微粒会很快被肝脏清除,只有几分钟的半衰期,一般不是动脉粥样硬化的主要原因。但是如果有APOE基因的某些变异,残体不能被肝脏快速清除,由残体运载的胆固醇一样会造成动脉粥样硬化。除此以外来自肠道的乳糜蛋白微粒因为特别大特别轻很难停留在分岔和转弯的位置,与动脉粥样硬化的关系很小。确实从基因的角度看,LDLR, APOB,PCSK9基因变异会降低肝脏回收LDL的能力,造成更多的LDL滞流在血液中,增加粘附到破损的血管壁上的机会。同理,如果APOE基因发生变异造成残体微粒不能被肝脏及时清除,从食品中吸收来的胆固醇就会较长时间滞留在血液中,也会增加进入血管壁夹层的机会从而增加动脉粥样硬化的风险。
第二章中我们讲过,LDL微粒都起源于肝脏,由LDL微粒运载的胆固醇称为低密度胆固醇LDL-C,就是我们常说的 “坏“胆固醇。“坏”胆固醇并不是这些胆固醇分子本身坏,而是LDL微粒更容易粘附到血管壁损伤处,并因此将LDL-C带入血管壁夹层,造成动脉粥样硬化。
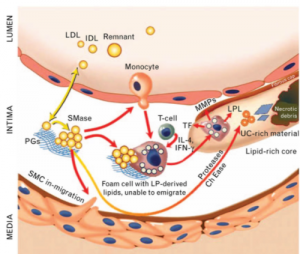
图5. 动脉粥样化硬化的病理过程示意图。富含胆固醇的脂蛋白微粒(LDL, IDL and remnant)进入到内皮细胞与平滑肌细胞之间的夹层,被糖蛋白粘附,被自由基氧化,触发免疫单核细胞浸入,泡沫状细胞形成,最后血管壁夹层中形成很复杂的油脂核心。
LDL微粒进入血管壁夹层后,少数还会跑出来重新回到血液循环中,而大多数没有跑出来的,会被夹层基质中带负电荷的蛋白多糖吸附,滞留在夹层中。蛋白多糖对LDL微粒的吸附多半是通过apoB100上带正电荷的区域。每一个LDL微粒都含有一个apoB100蛋白质,apoB100是身体中最大的蛋白质分子之一,有四千多个胺基酸,整个分子包裹在LDL微粒的表面,其肽链的N端有一个含正电荷的LDL受体结合区域,apoB100上这个区域就像”钥匙”一样,用于与细胞膜表面的LDLR受体结合,从而将LDL微粒转运进入肝脏和其他需要胆固醇的目标细胞。血管壁夹层基质含有大量蛋白多糖,在生理条件下这些蛋白多糖带负电荷,因此正负电荷相互吸引,LDL微粒一旦进了夹层就很难跑出来。时间一长,LDL上的胆固醇和油脂以及蛋白质都会被自由基氧化,成为oxLDL。被氧化了的胆固醇会被免疫系统视为有害物质,因此免疫细胞会前来清除。前来清除oxLDL的免疫细胞主要是巨噬细胞,通过吞噬机制把oxLDL粒吞进溶酶体中,在那儿LDL被消化拆分,部份油脂和胆固醇分子会被送到线粒体中燃烧产生能量,更多的胆固醇分子则会被酯化后贮存在巨噬细胞中的油泡里,如果夹层中积有太多的胆固醇和油脂,巨噬细胞会因为不断吞入胆固醇和油脂造成细胞中的油泡变得越来越大,巨大的乳白色的油泡在显微镜下看起来像泡沫一样,所以称为泡沫细胞。泡沫细胞的形成是动脉粥样硬化的一个里程碑。
这里要单独讲一下Lp(a),它是LDL的脂蛋白微粒的亚型,比普通的LDL多了一条尾巴,血脂查测时LDL-C浓度包括了Lp(a)上的胆固醇。每个Lp(a)是由一个LDL微粒通过二硫键与一条脂蛋白a肽链连接在一起。Lp(a)不像普通的LDL会被肝细胞上的LDLR受体结合并转运进入肝脏被清除,所以降胆固醇他汀类药不能降Lp(a)这部份胆固醇。更重要的是,Lp(a)的脂蛋白a肽链上有一个赖胺酸结合位点,使它比普通的LDL更容易粘附到血管壁损伤处,因此Lp(a)升高比普通的LDL升高危害更大。Lp(a)与心血管病关系极大,有很长的故事要讲,作者将在第四章遗传组合中详细介绍。
在清扫胆固醇的路上
血管壁夹层中的胆固醇是怎么被清除的呢?清除组织和细胞中多余的胆固醇主要依靠胆固醇反转应机制(reverse cholesterol transport )。胆固醇反转运比较复杂,主要由HDL来完成。HDL又称为高密度脂蛋白,专门负责去到身体各组织器官回收过剩或损伤的胆固醇,并将它们运回到肝脏加以再利用或消毁。我们可以将HDL想象成回收胆固醇的小车,其车身是apoA脂蛋白。apoA首先由肝脏合成并分泌到血液中,最初始的HDL只是裸体的apoA蛋白质,就像还没载货的空车。HDL随血液循环去到外周组织和细胞间,在那儿HDL会嵌合到细胞膜上的分子泵ABCA1上,诱导ABCA1将细胞内的自由胆固醇分子泵出到HDL上。到了HDL上的自由胆固醇分子然后被酯化后包装成紧密的胆固醇核,这样每个HDL微粒可以装载更多的胆固醇。催化胆固醇酯化的酶称为卵磷脂胆-胆固醇酰基转移酶(Lecithin Cholesterol Acyltransferase,LCAT),酯化反应需要卵磷酯作供体,蛋黄是卵磷酯含量最高的食物,所以吃鸡蛋会使HDL-C增高。在血液循环中,载有回收来的胆固醇的HDL微粒会与主要是甘油三酯的VLDL微粒碰头,这两个脂蛋白微粒之间会进行甘油三酯-胆固醇互换,这个交换过程由CETP基因负责;如果CETP活性不足,HDL上的胆固醇不能交换去到VLDL上,结果血液中高密度胆固醇HDL-C会升高。HDL-C就是我们通常说的“好”胆固醇。然而由于CETP失活而造成的HDL-C升高是否会有利于健康还不完全清楚,药物开发上通过药物抑制CETP确实能显著提高HDL-C,但这样升高的HDL-C在临床实验中并没有显示降低心血管病的收益,相反在好几个临床实验中显示增大风险。
胆固醇反转运的最后一步,是肝脏接收HDL运载回来的胆固醇,并把多余和受损的胆固醇转化为胆酸,以胆汁的形式排除肝脏。肝细胞用于回收HDL-C的受体SCARB1又称为清道夫受体(scavenger receptor),负责胆固醇反转运的最后一步。SCARB1负责卸载HDL微粒上的胆固醇,卸完货的空车HDL可以重新回到血液循环中再次装货,如果SCARB1基因发生变异,卸货不完全不及时,会使HDL微粒上胆固醇超载。比较常见的SCARB1 变异会造成HDL运载的胆固醇不能被肝脏有效回收,致使HDL上胆固醇超栽。携带这个SCARB1变异的人,会有非常高的HDL-C水平,通常在70-150mg/d L。请注意SCARB1变异造成的HDL-C升高不但不是好亊,反而严重增加心血管病风险。这是因为SCARB1缺陷虽然表面上看使血液中HDL-C升高,但实质上是卸空HDL出了问题,胆固醇反转运通道受阻。有SCARB1缺陷的人血液中HDL微粒会变得很大,可以比普通人的大出好几倍,心血管疾病风险也随之比普通人群高出2-3倍。SCARB1导致的HDL-C升高和心血管疾病风险只有通过基因检测才能确定。
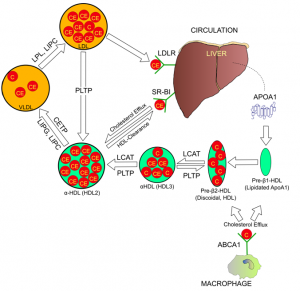
图6. 胆固醇反转运通道上HDL微粒从肝脏出发再回到肝脏的过程。各个基因在这条清扫胆固醇路上的作用
另外SCARB1基因除了在肝脏大量表达外,也在合成甾体激素的组织中有高表达,例如肾上腺,睾丸和输卵管等组织都表达大量的SCARB1。这表明由HDL收集来的胆固醇除了去肝脏外,也可被这些组织利用作为原料合成肾上腺皮质激素和性激素。
胆固醇反转运通道上的基因突变会影响动脉粥样硬化的进程。但这些基因的作用又远小于家族性高胆固醇血症基因。APOA1,ABCA1 和LCAT基因突变都会造成HDL-C水平极低。ABCA1缺失会造成称为丹吉尔病(Tangir’s disease),表现为HDL-C极度低,小于20mg/dL, 心血管病增加,但发病年龄在60-70岁以上。LCAT基因突变会导致鱼眼病(Fish eye disease),因为胆固醇不能被清除而积聚在眼睛角膜上让角膜看起来发白。CETP基因活性降低会使HDL-C升高,现在更多的证据指向如此升高的HDL-C并不降低心血管病风险。SCARB1基因突变会造成HDL-C显著升高同时心血管病风险也升高。所以HDL-C不能简单的理解成”好”胆固醇,HDL-C水平低固然不好,说明胆固醇反转运不给力,然而也不是HDL-C越高越好,要看是什么原因造成的升高。如果HDL-C特别高,超过70mg/dL就不见得是好亊了,最好能通过基因检测排出胆固醇反转运通道上的基因突变。
氧化自由基
血液中的过氧化自由基会直接损伤血管壁内皮细胞因此增加动脉粥样硬化。吸烟是已知的心血管疾病风险因素,吸烟的一个要害是直接增加血液循环中的氧化自由基,造成内皮细胞易受损。身体中的氧化自由基通常通过抗氧化剂和抗氧化酶来清除。在血管壁内皮细胞上通过表达超氧化物歧化酶(superoxide dismutase,SOD)来分解超氧化物,保护血管壁内膜。SOD3是一种存在于血管壁内皮细胞外的抗氧化酶,这个基因上某些变异会造成这个酶从内皮细胞表面脱落,结果血管壁内膜更容易被抗氧化自由基损伤。携带某些SOD3基因变异的人群患上心血管病的几率比没有这个突变的人大约要高1.7倍,而那些既有SOD3变异又吸烟的人,心血管病风险会更高。另外一个参与清除氧化胆固醇ox-LDL的基因OLR1,某些变异会增加大约二倍的独立风险。ox-LDL的形成是动脉粥样硬化的重要一环,ox-LDL会触发免疫单核细胞浸润进血管壁夹层,引发痰症反应,加速动脉粥样硬化的进程。因为胆固醇是脂溶分子,需要脂溶性抗氧化剂来保护。维生素E是身体中最常见的脂溶性抗氧化剂,但是一分子维生素E只能消灭一分子氧化自由基,而氧化自由基是源源不断的从细胞中产生,所以实际情况是身体中强大的抗氧化系统通过维生素C不断还原再生出有抗氧化活性的维生素E。维生素C负责还原维生素E,而维生素C本身可以被谷胱甘肽还原,谷胱甘肽被NADPH还原,而NADPH可以通过戊糖磷酸通路(Pentose phosphate pathway)分解葡萄糖源源不断的生产。想了解身体如何抗击氧化自由基的读者可以参考作者曾经写过的科普”维生素生素C的故事”。
另外如果身体缺乏叶酸,血液中同型半胱胺酸增高,也会增加内皮细胞损伤;可以造成同型半胱升高的基因突变,例如CBS基因的某些突变也会显著增加心血管病风险。多吃绿叶蔬菜或补充叶酸可以非常有效的降低同型半胱氨酸。
没人能够逃脱的动脉粥样
通过CT扫描可以检查和跟踪病人血管上粥样硬化斑块的大小和发展。这项研究发现,粥样硬化斑块区域的大小与年龄相关性最强,年龄可以解释大约50%原因,其次是性别,男性比女性的情况坏很多,然后分别是吸烟,舒张压高,在吃降血脂药(有高血脂)和在吃降血压药 (有高血压)。跟踪一年后这些病人的斑块进展情发现,LDL-C高是斑块继续变大变坏的第一大因素,高血压是第二大因素,高甘油三酯是第三大因素,但比前面两个因素的作用小很多,其他因素影响甚微。
我们可以清淅的看到动脉粥样硬化的发生和发展是一个长期累积的过程,我们出生就开始一点点沉积病变,但要到了中老年后才会出现症状。只要我们活着,只要血液在我们身上流淌,我们就无法逃脱动脉粥样硬化的命运。有的人运气好天生有最佳的基因组合,又有良好的饮食健康习惯,活到100岁动脉血管仍然可以比较畅通;而有的人很不幸天生基因组合就不够好,再加上饮食生活习惯选择与基因不对应让情况更糟,就有可能在正当壮年时发生心肌梗塞亊件。没有人能选择自己的基因组合,都是父母那儿遗传来的,但是我们可以选择管理好我们的基因,争取早诊早预防早治疗,利用科学的手段改变命运。
且听下回分解
从基因的角度解析心血管疾病风险
(一)从肝脏到心脏
(二)细说胆固醇
(三)没人能逃避的动脉粥样硬化
(四)遗传组合
(五)精准医学与营养学
参考文献:
1. https://en.wikipedia.org/wiki/Atherosclerosis
2. https://en.wikipedia.org/wiki/Cardiovascular_disease
3. https://www.heart.org/en/health-topics/cholesterol/about-cholesterol/atherosclerosis
4. https://www.merckmanuals.com/home/heart-and-blood-vessel-disorders/biology-of-the-heart-and-blood-vessels/biology-of-the-blood-vessels
5. Lee D, Chiu J. Atherosclerosis and flow: roles of epigenetic modulation in vascular endothelium. J Biomed Sci. 2019 Aug 7;26(1):56.
6. Borén J, Kevin Jon Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016 Oct;27(5):473-83. PMID: 27472409.
7. Tansey EA, Laura E A Montgomery LEA, Quinn JG, Roe SM, Johnson CD. Understanding basic vein physiology and venous blood pressure through simple physical assessments. Adv Physiol Educ. 2019 Sep 1;43(3):423-429.
8. Wobst J, Schunkert H, Kessler T. Genetic alterations in the NO-cGMP pathway and cardiovascular risk. Nitric Oxide. 2018 Jun 1;76:105-112. PMID: 29601927.
9. Kessler T, Wolf B, Niclas Eriksson N, et al. Association of the coronary artery disease risk gene GUCY1A3 with ischaemic events after coronary intervention.
10. Nordestgaard BG and Benn M. Genetic testing for familial hypercholesterolaemia is essential in individuals with high LDL cholesterol: who does it in the world? Eur Heart J. 2017 May 21;38(20):1580-1583. PMID: 28419271
11. Cardiovasc Res. 2019 Aug 1;115(10):1512-1518. PMID: 30768153Bowe B, Xie Y, Xian H, Balasubramanian S, et al. High Density Lipoprotein Cholesterol and the Risk of All-Cause Mortality among U.S. Veterans. Clin J Am Soc Nephrol. 2016 Oct 7;11(10):1784-1793. PMID: 27515591.
12. Madsen CM, Anette Varbo A, Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur Heart J. 2017 Aug 21;38(32):2478-2486. PMID: 28419274.
13. Zanoni P, Sumeet A Khetarpal SA, Daniel B Larach DB et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016 Mar 11;351(6278):1166-71. PMID: 26965621.
14. Tietjen I, Hovingh GK, Singaraja R, et al. Increased risk of coronary artery disease in Caucasians with extremely low HDL cholesterol due to mutations in ABCA1, APOA1, and LCAT. Biochim Biophys Acta. 2012 Mar;1821(3):416-24. PMID: 21875686.
15. Frikke-Schmidt R, Nordestgaard BG, Stene MC, et al. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008 Jun 4;299(21):2524-32.
16. Tatsuguchi M,Furutani M, Jun-ichi Hinagata J,et al. Oxidized LDL receptor gene (OLR1) is associated with the risk of myocardial infarction. Biochem Biophys Res Commun. 2003 Mar 28;303(1):247-50. PMID: 12646194.
17. J. David Spence. Genetics of atherosclerosis: the power of plaque burden and progression: invited commentary on Dong C, Beecham A, Wang L, Blanton SH, Rundek T, Sacco RL. Follow-Up association study of linkage regions reveals multiple candidate genes for carotid plaque in Dominicans atherosclerosis 223 (1) (2012) 177-183.. 2012 Jul;223(1):98-101. PMID: 22648086.
★更多深度解析访问《美中药源》~
★ 请关注《美中药源》微信公众号 ★


:何为生物类似药?")


:生物类似药 VS. 化学仿制药")



- 路人丙: 新药发现的低悬果实
- Pipi_WHU: 新药发现的低悬果实
- 路人丙: Galapagos放弃IPF资产、吉利德空手而归
- Tan, Chengfang: Galapagos放弃IPF资产、吉利德空手而归
- 配体效率遭到质疑 | 美中药源: 你笑我无知类药性本不存在?
- Songdanqing: 你笑我无知类药性本不存在?
- 路人丙: 中国新药的新时代
- 康, 荣明: 中国新药的新时代
合作伙伴









 微信号:美中药源
微信号:美中药源