
从基因的角度解析心血管疾病风险(四)遗传组合
By 沈骊 GB HealthWatch

第四章 遗传组合
我们经常看到这样的例子,父母都非常健康长寿,但其中的一个子女却在相对年轻的时候就发生了心血管疾病。或者是祖父母那一辈有过心血管病史,好像隔代遗传到了自己这一辈。一般来说,如果父母中的一方很年轻时就发生过心脑血管疾病,子女的风险也会增高;如果父母都不曾被诊断过心脑血管疾病,子女的风险也会相应降低。但是这里说的是相对风险,具体对每一个体来说,绝对风险是由父母那儿遗传来的确切基因组合决定的。这有点像漂亮的父母多半会生出漂亮的孩子来,但没有绝对保证。
作者见证过一名十四岁就发生脑卒中的少年的DNA样本。脑卒中是动脉粥样硬化造成脑局部缺血引起的。这名不幸的少年从母亲处遗传了会造成高胆固醇血症的LDLR致病突变和APOE4变异,又从父亲处遗传了造成高Lp(a)的LPA短型变异。这种最差组合让这名少年的血液中除了低密度胆固醇LDL-C极高外同时Lp(a)也非常高,这两个血液指标分别是独立的也是最大的两个促使动脉粥样硬化形成的原因。然而,这位少年的父母本人都没有心脑血管病症状。基因检测发现,母亲虽然携带着与儿子相同的LDLR致病突变和APOE4变异,但她的LDL-C只是中等程度增高,原因是这位母亲是APOE2/E4杂合子,APOE2变异可以显著降低胆固醇和Lp(a),而APOE4变异会增加; 同时这位母亲还携带会降低胆固醇的ANGPT3基因变异,这两个有保护性的基因变异恰恰没有遗传给儿子;而父亲那边也携带着与儿子同样的会造成Lp(a)升高的短型LPA变异,但父亲是APOE2/E3杂合子,APOE2变异会使胆固醇和Lp(a)都降低,这个有保护作用的APOE2变异也恰恰没有遗传给儿子;儿子刚好从父母那儿遗传到了最坏的基因组合:LDLR致病突变+LPA短型变异+APOE4变异。继续追踪发现,这名少年的姨妈(母亲的姐姐)有高胆固醇和心血管病,但是很晚才发病。基因检测揭示姨妈也携帶有和她母亲一样的LDLR致病突变,但是没有她母亲的两个保护性变异,所以得病风险比她母亲高。与他姨妈相比,这名少年又从父亲那儿遗传了额处的有严重致病风险的LPA短型变异,这个最差基因组合让他在如此年轻的年龄就发生了脑卒中亊件。这个一病例让我们看到虽然子女的基因是从父母来的,但不能完全依据父母有无心脑血管病史来推测子女的得病的风险,子女可能得到父母的好基因组合也可能得到他们的坏基因组合。
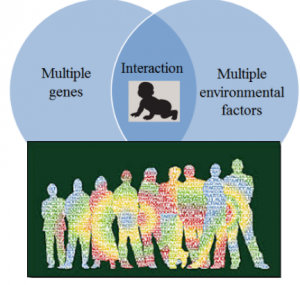
图1. 心血管疾病属于复杂多基因疾病,由多个遗传因子和多种环境因素相互作用最终决定得病风险
心血管疾病属于复杂型多基因疾病,这类疾病的遗传成份复杂,由多个遗传因子相互作用而决定,同时饮食营养,生活习惯,体育锻炼等会与遗传因素相互作用而改变得病风险。多基因疾病不属于经典的遗传病,很难用单个基因的突变来解释得病的风险,既不属于隐性遗传,也不属于显性遗传,属于多基因风险组合遗传。参与风险决策的基因涉及多个生物通道,包括胆固醇的运送回收和反转运通道,血压和血管紧张度的调节,抗氧化自由基通道,炎症反应,血栓形成等。遗传风险是由来自父母双方的多个基因决定,而且增加风险的变异有时可以被降纸风险的变异中和。 第一,有的人恰好幸运,遗传了父母基因的最佳组合,即便有心脑血管疾病家族史,自己可能完全没亊;而有的人刚好倒霉,遗传了父母基因的最差组合,即便父母从来没有心脑血管病史,自己也可能才人到中年就发生心血管病事件。第二,即便在相等的遗传风险下,饮食营养和生活习惯可能会很大程度改变得病的年龄和轻重。作为中国人,我们的父母辈曾经很长时间生活在食品缺乏的环境里,一生中大鱼大肉吃得较少,而青菜豆腐稀饭吃更多,在这样的饮食环境下,他们的血脂血压和血糖总体比我们这一辈人低,因此患心血管疾病的年龄也可能会向后推迟几年。第三,多基因高风险变异可以隔代组合。因为是多基因病,某个基因增加的风险可能被另一个基因抵消,如果一个人同时携带等量的增加风险和降低风险的变异,这个人就可能完全没有临床症状,但是仍然可以将风险变异传给下一代。比如祖辈有明显心血管病史,而父辈没有心血管病史,但是这不能肯定父辈没有遗传到祖辈的风险变异,而有一种可能是风险变异造成的危害被另一个起保护作用的变异抵消了;到了子辈,子辈可以从父辈那儿遗传到了来自祖辈的风险变异,却没有遗传到起保护作用的变异,更差的组合还可能是遗传到父母两个家族所有风险变异的最差组合。
坏基因组合,好基因组合
最常见的最差基因组合应该是家族性高胆固醇血症LDLR突变+LPA短型变异。大约1/200的人携带有家族性高胆固醇血症基因突变,这些人发生心血管事件的几率大约是没有携带突变者的13-20倍,如果这些人同时还携带LPA短型变异,那么发生心血管病事件的几率会在原来基础上再增加二倍,同时发病时间还会再提早4-5年。LPA短型变异很常见,大约20%的白人,30%的非洲人和6%的亚洲人携带LPA短型变异。只要携带一条LPA短型变异,就会使血液中Lp(a)升高。有这种组合的人表现为LDL-C很高,Lp(a)也很高。
其次,家族性高胆固醇血症基因突变与胆固醇反转运通道上的任何基因突变组合在一起,例如ABCA1,APOA1,LCAT和SCARB1等。也会进一步增加心脑血管病的发病几率同时使发病年龄提前。但是固醇反转运基因突变远没有LPA短型变异那么常见,所以这种组合虽然也很坏但是并不常见。有这种组合的人表现为LDL-C很高,HDL-C很低或很高。
隐型杀手
LPA短型变异与SCARB1失活变异的组合也是很坏的组合,而且是隐性杀手。SCARB1又称为清道夫受体,负责胆固醇反转运的最后一步,吸收由HDL收集来的胆固醇HDL-C。某些SCARB1基因变异会造成胆固醇反转运通道受阻,结果血液中HDL-C水平会显著升高,同时伴随心血管病风险也增加。最近有研究发现,SCARB1至少部分参与Lp(a)的清除,如果SCARB1失活,血液中的Lp(a)会进一步升高。因为常规体检中并不检测Lp(a),Lp(a)升高只会表现为中轻度LDL-C升高,而SCARB1失活会表现为HDL-C升高,因此有这种基因组合的人,血脂检查报告完全看不到问题。一般情况下我们会认为HDL-C是”好”胆固醇,所以越高越好,很难会想到HDL-C高有可能是胆固反转进通道出了问题。由于Lp(a)升高造成的LDL-C中轻度升高也不太会引起注意,常用的LDL-C/HDL-C比例仍然很好,基本上是完美的血脂报告。如果没有发生心脑血管病事件,完全沒有人会想到有这么好的血脂检查报告的人其实有很高风险。这种基因组合在人群中应该相对常见,大约20%的人有LPA短型变,2%的人有一个已知的SCARB1失活变异。如果一个人血脂检测报告近乎完美,无不良生活习性,却在五六十岁的年龄就突发心肌梗塞亊件,可能就要怀疑有这种隐性杀手基因组合了。
好基因组合
与心血管病风险相关的好几基因都与长寿有关。最常见的是APOE2变异,这个变异在人群中很常见,亚洲人中大概20%的人携带这个变异。有单个拷贝APOE2变异的人的最大特征是血液中胆固醇水平较低,同时APOE2也会降低血液中Lp(a)水平,高胆固醇和高Lp(a)是两个最大的心血管疾病风险,因此可见遗传到APOE2变异的双重好处。另外与APOE4相反,APOE2变异也降低老年痴呆风险。但并不是APOE2变异越多越好,有两个拷贝APOE2/E2变异的人有20-50%风险会患上三型高血脂症,表现为甘油三脂严重升高,总胆固醇升高,LDL-C正常或微弱升高,心血管病风险增加5-10倍。不过有APOE2/E2的人数很少,在人群中不大于1%。
另一个著名的长寿基因变异是APOA1的米兰变异,这个变异在意大利的一个长寿村发现,有制药公司还曾模仿这个变异设计了一个多肽药物用于治疗动脉粥样硬化,但是药物最终没有上市。
我们中的大多数人会在最差与最佳组合之间,从父母那儿遗传到多个中等风险程度的变异同时也遗传到一些有保护作用的变异。因为基因组合的不同再加上饮食营养和生活方式的参与,有的人早到20多岁的年龄就可能发生第一次心血管病事件,有的人可能晚到100多岁才出问题,而我们中的大多数人则在他们之间。
家族性高胆固醇血症基因和LPA基因短型变异是两个最大的心脑血管疾病的遗传因素,我们将在下面详细介绍。
基因和家族性高胆固醇症
家族性高胆固醇血症(famila hypercholesterolemia,FH) 是目前研究得最多,致病机理最清楚,与心血管病相关性最强的遗传病。主要症状为血液中低密度胆固醇(LDL-C)浓度很高,而且是从年轻时侯就开始高。LDL-C就是我们常说的”坏胆固醇”。在没有使用降血脂药物情况下,LDL-C超过190mg/dL(4.9 mmol/L),并且有心血管疾病家族史,就被临床诊断为FH。基因检测是确诊FH的黄金标准(gold standard)。如果FH没有被早期诊断和治疗,男性在55岁以前,女性在60岁以前发生心肌梗塞或者脑卒中的几率比一般人高10-20倍。很愦撼,即使在医疗发达的美国,90%以上的FH仍然没有被诊断和治疗。绝大多数FH是在心肌梗塞事件发生后才被诊断。然而,早发心肌梗塞是可以预防的,通过基因检测早期确诊的FH患者,从青少年时期就开始采用降胆固醇药物治疗,可以推迟心血管病发病时间10-15年。

图2. 家族性高胆固醇血症(FH)是显性遗传,如果一个人有FH,这个人的孩子50%的机会也有FH
家族性高胆固醇血症分为单基因型(mongenic FH)和多基因型(polygenic FH)。一般而言,单基因FH患者比多基因FH患者胆固醇水平更高,会在更年轻的时候发生心肌梗塞或其他心脑血管病。如果年龄小于二十岁的年轻人胆固醇水平就很高,是单基因FH的可能性就很高。单基因FH是指一个基因的突变就足于导致疾病症状。单基因FH被认为是显性遗传,也就是说只需要携带一个拷贝的致病突变,就会出现高胆固醇。父母双方中只要一方有单基因FH,子女就有50%的机率遗传到这种病。绝大多数单基因FH由三个基因造成,最常见的是LDLR基因突变,90%以上的单基因FH都是LDLR突变,5% 来源于APOB基因突变,1%来自于PCSK9基因突变,还有极少数的病例由LDLRAP1和STAP1 等基因突变造成。在FH中,有些患者同时携带两个拷贝致病突变(homozygous FH),这些患者的胆固醇浓度会非常非常高,如果没有被极时诊断和采取极积的降胆固醇治疗,可能在二十几岁的年龄就会发生缺血性脑卒中或心肌梗塞。
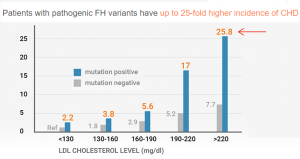
图3. 家族性高胆固醇血症 (FH)患者的胆固醇水平与对应的心血管病风险
LDLR是在肝脏和其他需要胆固醇的组织上表达的一种LDL脂蛋白受体,主要功能是与血液中运载胆固醇的低密度脂蛋白微粒LDL结合,通过受体介导的细胞内吞机制,将胆固醇转运进入肝脏和目标组织。LDLR是一个特别容易发生突变的基因,已经在人群中检测到超过1200种突变,其中有些突变是致病的,会使LDLR转运胆固醇的功能全部或部分丧失,因此造成不同程度的高胆固醇。除了点突变外,LDLR基因大片断删除或插入也比较常见,大约3%的LDLR致病突变是外显子删除,这些突变造成的LDL-C升高也是往往更加严重。在欧洲和日本人中都发现不少外显子删除突变,在中国人中未见报道,可能是还没有人研究过。通过插样估计,中国太约有三百八十万人携带FH致病突变,但是已经诊断的人数很少,说明大多数没有被正确诊断。总之LDLR是一个突变特别多的基因,在我们做过基因检测的FH病人中,几乎每个家族都有各自特殊的突变位点,很难见到重复。所以FH突变必须用二代DNA测序来检查,像23andMe那样的基因芯片对检测FH突变几乎没有用。
APOB基因变异导致的FH症状较轻。只有很少几个APOB基因上的突变会导致FH, 这些突变都发生在受体结合区域(LDLR-binding domain), 结果会造成APOB编码的蛋白apoB不能很好的与LDLR结合, 减慢LDL微粒被肝脏回收。APOB基因上的大多数变异并不导致FH, 相反,这些变异导致低血脂症(hypolipidemia)。同样,大多数PCSK9基因变异导致低血脂症,只有很少几个造成PCSK9活性增加的变异会导致FH。PCSK9是一个酶控LDLR的降解和重复使用。PCSK9活性升至高,LDLR被降解增加,PCSK9失去活性,LDLR不被降解而被循环回到细胞膜表面再次使用。这也是PCSK9抑制剂可以降胆固醇的原理。
这里可以看出,三个FH基因表面上看起来生物功能完全不同,一个是细胞膜上的受体,另一个是组成脂蛋白微粒的核心蛋白,最后一个是蛋白酶,但是这三个基因的突变都会影响LDLR转运胆固醇的能力,最终影响肝脏回收血液中胆固醇的能力。如果肝脏不能有效回收胆固醇,血脂就会升高,动脉粥样硬化就会加速,心脑血管病风险就随之增加。
Lp(a)的故事
Lp(a)发音叫L P little a,它是LDL脂蛋白微粒的一个亚型,比普通的LDL多了一条尾巴。每个Lp(a)是由一个LDL微粒通过二硫键与一条脂蛋白a肽链连接在一起。Lp(a)不像普通的LDL会被肝细胞上的LDLR受体结合并转运进入肝脏被清除,所以降胆固醇他汀类药不能降在Lp(a)里的这部份胆固醇。更重要的是,Lp(a)的脂蛋白a肽链上有一个赖胺酸结合位点,使它比普通的LDL更容易粘附到血管壁损伤处,因此Lp(a)升高比普通的LDL升高危害更大。血液中Lp(a)升高增加大约两倍的独立心血管病风险,同时也提早发病时间。
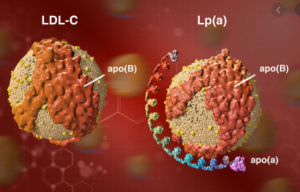
图4. Lp(a) 是一种LDL微粒的亚型,Lp(a)像多了一条尾巴的LDL。左边图示LDL微粒,右边图示Lp(a)微粒。
Lp(a)微粒上的脂蛋白a肽链由LPA基因编码。LPA是一个在进化树上很晚才出现的基因,这个基因仅仅出现在人类和人类的祖先灵长类以及其他很少几种动物中,例如豚鼠。因为灵长类动物和豚鼠也是少数几种在进化过程中丧失了合成维生素C基因的动物,LPA基因的出现与失去合成维生素C基因的物种相重合。据此大科学家鲍林曾经提出一个关于心血管病起源的进化假说:人类因为在进化过程中失去了自主合成维生素C的酶,当维生素C供应不足时,例如冰川时期长期没有新鲜植物和水果吃,灵长类动物就会因为缺乏维生素C得坏血病死亡;在面对物种生死存亡的时刻,可能是一次偶然的DNA复制错误,PLG基因的部分片断被插入到染色体中形成了LPA基因。LPA基因编码的脂蛋白a一边能与血液中的LDL微粒通过二硫键结合,另一边又能与血管破损处暴露出来的赖胺酸结合,从而可以将血液中的胆固醇粘附到血管壁的破损处,起到了修补血管壁防止出血的作用。LPA基因可能拯救了灵长类动物在冰川世纪没有因为出血死光,但是添加了这个LPA基因后人类从此增加了动脉粥样硬化的风险。豚鼠是另一个在进化上失去维生素C活成酶同时获得了LPA基因的动物。其他我们熟悉的动物像鼠兔猫狗牛马猪羊统统都能自主合成维生素C,也统统都没有LPA这个基因。
LPA基因是通过复制删切纤维蛋白溶酶原基因PLG衍生出而来,它保留了部分PLG片段同时完全删除了编码酶活性的部份,然后其中第四个外显子又自身重复成为多个拷贝称为Kringle IV repeat(KIV2)。LPA基因所含的KIV2拷贝数目不同造成了LPA基因的长短多态性,最短的LPA基因只含2个拷贝KIV2,而最长的超过40个拷贝。人群中存在各种长度的LPA基因,几乎是呈正态分布。非洲人群携带较短的LPA比较多,亚洲人群携带较长的LPA比较多。LPA基因的长短决定了血液中Lp(a)浓度的高低,LPA基因越短,Lp(a)浓度就越高,出现动脉粥样硬化越早。鲍林假设认为人类之所以会患动脉粥样硬化是进化过程中两害相权取其轻的结果。进化过程选择性保留了LPA基因让人类的祖先灵长类动物在严重缺乏维生素C的环境下没有因为出血而死亡,幸存下来的灵长类动物因此保留了LPA基因。作者认为LPA基因在进化的早期都是短型的,会产生很高的Lp(a),防止出血的效果很好,但是太容易造成早发动脉粥样硬化和心肌梗塞了。随后的进化过程中,冰川纪结束,灵长类动物又能获得足够的维生素C了,因此不会因为坏血病而死亡,也就不需要LPA基因所产生的Lp(a)了。这时那些复制过程中偶然增加KIV2拷贝数目的LPA基因变异有了进化选择优势,那些获得较长LPA基因的人不会年纪轻轻就死于心血管病了,寿命变得更长,这就形成了今天我们观察到的LPA基因有长有短的多态性。总体来说亚洲人有长LPA变异的人最多,非洲人有短LPA的人最多,这也可能是亚洲人寿命相对其他种类来说较长的原因。
LPA基因特别长的人身上几乎测不到Lp(a),这些人得心血管疾病的风险也相对低。LPA基因的变长有可能是进化过程在选择性的降低Lp(a)的负面作用。Lp(a)的浓度几乎完由LPA基因的长短决定,基因越短,浓度越高。基本不受饮食和锻练改变,目前也没有药物可以降低Lp(a)水平。而且每个人的两条LPA基因通常也是不一样长,血液中的Lp(a)浓度主要由短的那条LPA基因决定。Lp(a)的真实生理功能并不清楚,人和人之间Lp(a)浓度差距可以高达千倍,在有些人身上几乎一点都检测不到,而在另一些人身上Lp(a)可以高达200-300mg/dL,这暗示着我们的身体可能完成不需要Lp(a)的功能,至少是在现在人类生活的环境下。因为老鼠根本就没有LPA这个基因,所以无从做基因删除小鼠(knockout mouse)来验证LPA的功能。但现实生活中存在完全丧失LPA基因表达的人,虽然这些人的血液中完全检测不到Lp(a),但是他们都活得好好的没啥毛病,相反Lp(a)高的容易发生心肌梗塞。这从另一个侧面说明了LPA基因的产生只是进化过程中生物面对一种特殊生活环境的选择(例如冰川纪严重缺乏维生素C),这种特殊生活环境消失后,LPA基因真正用途也就不被需要了。
不过有一项日本的研究发现,Lp(a)特别低的人得脑溢血的风险会增加。我们前面讲过亚洲人群有LPA短型的人比欧美人群要少得多,而且有LPA长型的人要多得多,这就是说亚洲人群Lp(a)的浓度相对比欧美人要低。有一个事实很值特注意,亚洲人脑溢血的发病率大约是欧美人的4倍,Lp(a)低很有可能是一个原因。
参与动脉粥样硬化形成和心血管疾病风险的基因有将近100个,这里只是给大家建立一个概念,就不一一细述了。
且听下回分解
从基因的角度解析心血管疾病风险
(一)从肝脏到心脏
(二)细说胆固醇
(三)没人能逃避的动脉粥样硬化
(四)遗传组合
(五)精准医学与营养学
参考文献:
1. Nordestgaard BG and Benn M. Genetic testing for familial hypercholesterolaemia is essential in individuals with high LDL cholesterol: who does it in the world? Eur Heart J. 2017 May 21;38(20):1580-1583. PMID: 28419271.
2. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013 Dec;34(45):3478-90a.
3. Madsen CM, Anette Varbo A, Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur Heart J. 2017 Aug 21;38(32):2478-2486. PMID: 28419274.
4. Jiang L, Li-Yuan Sun LY, Yan-Fang Dai YF, et al. The distribution and characteristics of LDL receptor mutations in China: A systematic review. Sci Rep. 2015 Nov 26;5:17272. PMID: 26608663.
5. Shinohara M. Kanekiyo T, Tachibana M. APOE2 is associated with longevity independent of Alzheimer's disease. Elife. 2020 Oct 19;9:e62199. PMID: 33074098.
6. https://en.wikipedia.org/wiki/ApoA-1_Milano
7. Alexander ET, Tanaka M, Momoe Kono M, et al. Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009 Jul; 50(7): 1409–1419.
8. Zanoni P, Sumeet A Khetarpal SA, Daniel B Larach DB et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016 Mar 11;351(6278):1166-71. PMID: 26965621.
9. Tietjen I, Hovingh GK, Singaraja R, et al. Increased risk of coronary artery disease in Caucasians with extremely low HDL cholesterol due to mutations in ABCA1, APOA1, and LCAT. Biochim Biophys Acta. 2012 Mar;1821(3):416-24. PMID: 21875686.
10. Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57(8):1339-59.
11. Chasman DI, Dov Shiffman D, Zee RYL, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009 Apr;203(2):371-6. PMID: 18775538.
12. Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014 Feb 11;63(5):470-7. PMID: 24161338.
13. Emdin CA, Khera AV, Natarajan P, et al. Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J Am Coll Cardiol. 2016;68(25):2761-2772. PMID: 28007139
14. Paquette M, Bernard S, Thanassoulis G, Baass A. LPA genotype is associated with premature cardiovascular disease in familial hypercholesterolemia. J Clin Lipidol. 2019 Apr 23. pii: S1933-2874(19)30082-0. PMID: 31103339.
15. Scipione CA, McAiney JT, Simard DJ, et al. Characterization of the I4399M variant of apolipoprotein(a): implications for altered prothrombotic properties of lipoprotein(a). J Thromb Haemost. 2017 Sep;15(9):1834-1844.
16. Chasman DI, Shiffman D, Zee RY, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009 Apr;203(2):371-6. PMID: 18775538. (aspirin reduce CVD event about two fold but almost not effect to non-carrier).
17. Yang X, Sethi A, Yanek LR, et al. SCARB1 Gene Variants Are Associated With the Phenotype of Combined High High-Density Lipoprotein Cholesterol and High Lipoprotein (a). Circ Cardiovasc Genet. 2016 Oct;9(5):408-418. PMID: 27651445.
18. M Rath, L Pauling. Hypothesis: lipoprotein(a) is a surrogate for ascorbate. Proc Natl Acad Sci U S A. 1990 Aug;87(16):6204-7. PMID: 2143582.
19. Lawn RM, Wade DP, Hammer RE, Chiesa G, et al. Atherogenesis in transgenic mice expressing human apolipoprotein(a). Nature. 1992 Dec 17;360(6405):670-2. PMID: 1465128.
20. Langsted A, Kamstrup PR, Nordestgaard BG. High Lipoprotein(a) and Low Risk of Major Bleeding in Brain and Airways in the General Population: a Mendelian Randomization Study. Clin Chem. 2017 Nov;63(11):1714-1723. PMID: 28877919.
21. Ishikawa S, Kotani K, Kario K,. Inverse association between serum lipoprotein(a) and cerebral hemorrhage in the Japanese population. Thromb Res. 2013 Feb;131(2):e54-8. PMID: 23260441.
22. Shen AY, Yao JF, Somjot S Brar SS, et al. Racial/ethnic differences in the risk of intracranial hemorrhage among patients with atrial fibrillation. J Am Coll Cardiol. 2007 Jul 24;50(4):309-15. PMID: 17659197.
★更多深度解析访问《美中药源》~
★ 请关注《美中药源》微信公众号 ★


:何为生物类似药?")


:生物类似药 VS. 化学仿制药")



- 路人丙: 新药发现的低悬果实
- Pipi_WHU: 新药发现的低悬果实
- 路人丙: Galapagos放弃IPF资产、吉利德空手而归
- Tan, Chengfang: Galapagos放弃IPF资产、吉利德空手而归
- 配体效率遭到质疑 | 美中药源: 你笑我无知类药性本不存在?
- Songdanqing: 你笑我无知类药性本不存在?
- 路人丙: 中国新药的新时代
- 康, 荣明: 中国新药的新时代
合作伙伴









 微信号:美中药源
微信号:美中药源