
ADC到底是什么?(十四)ADC是正宗前药吗?

ADC一般被认为是一个前药(prodrug)。前药的定义是药物本身没有活性,在体内经代谢处理后才有活性的药物,如果药物和代谢产物都有活性那么严格讲不能称为前药。为什么要求前药激活前没有生物学活性呢?那是因为有些情况下你希望药物在疾病组织的特定环境下激活,减少对非疾病组织造成伤害。一个相关的设计思路是软药(soft drug,也叫逆代谢药物、retrometabolic drug),这种药物完成治疗任务会快速代谢失活、比如局麻药物。过时自合飘零去对于ADC毒素来说也是一个有益性质,因为毒素如果离开肿瘤被快速代谢失活会降低系统毒性。
ADC因为毒素安全窗口很小所以希望通过前药设计增加治疗窗口,但直到最近我们并没有确切数据证明ADC主要是通过游离毒素杀伤肿瘤的。小分子前药设计一般是在分子对活性敏感区引入代谢基团以保证前药无活性,但现在ADC设计中小分子毒素一般是在对结构变化不敏感区域通过链接子偶联到抗体上。比如常用的喜树碱类药物在反应中心羟基内酯的对面区域几乎任何结构改造都有TOP1抑制活性,进入临床的十几个TOP1抑制剂在这个区域改造千奇百怪。所以从与靶点结合角度看,毒素并不在意连在末端的是一个完整抗体、一个残缺的抗体、还是一个乙酰基,因为这个长尾巴并不打扰毒素与靶点的结合。
但药物除了能与靶点高效结合外还要进入靶点所在区域,所以ADC是不是前药取决于哪一级代谢产物、包括ADC本身能否在胞内遇到靶点。有研究发现把VC链接子用蛋白酶无法识别的非天然氨基酸替代、或敲除细胞组织蛋白酶从而改变胞内毒素存在形式,MMAE ADC的杀伤力并不会下降,但尚无直接证据ADC本身不经过任何降解就可以杀伤肿瘤细胞。一般认为抗体药物无法调控胞内机制(当然这个理念也在改变),给人印象抗体在胞内无所作为。但多数抗体不能调节胞内靶点是因为没有进入细胞的路径,ADC因为有胞外靶点帮忙可以进到胞内,所以有可能以完整抗体身份在胞内做一些事情。
抗体并非进入细胞就粉身碎骨,降解也需要时间。比如曲妥珠单抗在SK-BR-3细胞中的降解半衰期是16个小时,长期给药时的药物稳态期胞内可能有相当浓度的抗体药物。有研究显示不可裂解ADC药物T-DM1净内吞速度是胞内降解速度的~2倍,所以也可能在胞内蓄积一定ADC药物浓度。尽管内吞从机制上看是通过胞饮所以抗体可能被脂双层包裹,但是还是有可能通过各种机制与细胞质中靶点相遇。因为DM1靶点是细胞质中的微管,所以T-DM1有可能在被降解之前就开始破坏微管结构,那么T-DM1就不能称作真正意义的前药。
对于毒素为DNA损伤剂的ADC如T-DXd来说这种可能性要小很多,因为靶点在细胞核内、而抗体进入细胞核的难度比进入细胞质大很多。今年一三共的科学家用荧光标记T-DXd的抗体部分发现在HER2阳性肿瘤细胞体外实验中的确只有少量抗体(红色)能够到达细胞核(蓝色),多数被送到溶酶体(绿色,图1)。而在体内肿瘤实验中DXd(红色,图2)则可以进入细胞核(蓝色),这说明T-DXd是一个货真价实的前药。
当然最近也有研究说喜树碱类药物的靶点不仅是TOP1,也有其它蛋白靶点,因为无TOP1表达的细胞也对某些喜树碱衍生物敏感。这就不排除ADC可以直接参与靶点调控了,因为细胞质中存在大量DXd(图2白色)。这些DXd可能是游离的、也可能是T-DXd,此前在分辨率较低的荧光标记实验中发现DXd和曲妥珠基本是在同一区域出现、说明至少部分DXd信号来自T-DXd。
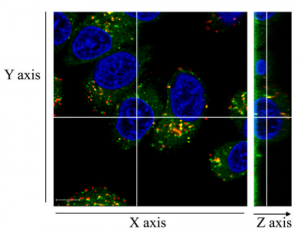
图1

图2
ADC血药浓度即使按摩尔浓度算也通常远高于毒素,但这主要与ADC、毒素的清除率和组织分布差异有关,不能说明肿瘤细胞内ADC原药也比游离毒素多很多,毕竟我们还希望肿瘤细胞能成为加工ADC、释放毒素的一个车间。这个数据通常是未知的,所以难以估计ADC直接参与细胞杀伤的程度。 当然前面讲了除了ADC原药浓度,毒素靶点在胞内地址决定了ADC是否需要“激活”才能起效。
ADC进入细胞后是先需要释放毒素再开始杀伤,还是可以一边切断链接子、同时可以开始抑制靶点活性可能与很多因素有关。如果是前面一种情况,链接子断裂效率会影响ADC疗效,ADC药物可能特异性更高、更安全。如果是后者,ADC可能会更广谱、当然也可能毒性更大。随着更多数据的产生我们对这些过程的认识也会越来越清晰,可以更有的放矢地设计新一代ADC。
美中药源原创文章,转载注明出处并添加超链接,商业用途需经书面授权。★更多深度解析访问《美中药源》~
★ 请关注《美中药源》微信公众号 ★


:何为生物类似药?")


:生物类似药 VS. 化学仿制药")



- 路人丙: 新药发现的低悬果实
- Pipi_WHU: 新药发现的低悬果实
- 路人丙: Galapagos放弃IPF资产、吉利德空手而归
- Tan, Chengfang: Galapagos放弃IPF资产、吉利德空手而归
- 配体效率遭到质疑 | 美中药源: 你笑我无知类药性本不存在?
- Songdanqing: 你笑我无知类药性本不存在?
- 路人丙: 中国新药的新时代
- 康, 荣明: 中国新药的新时代
合作伙伴









 微信号:美中药源
微信号:美中药源