
ADC到底是什么?(十六)善变的喜树碱
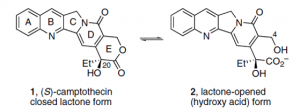
ADC自2000年首次批准已有二十几年历史,但真正火起来是2019年T-DXd的出现。传统的ADC设计自以为是魔法子弹枪法所以什么样的毒素都敢用,甚至是越毒越好、因为这样就不需要太高剂量抗体。但是我们此前已经讲过这个理念的诸多瑕疵,即使HER2这样高密度靶点、罗氏这样的肿瘤药开发高手如果子弹用错了也会让魔法失灵。小分子喜树碱衍生物已有两个上市药物(伊利替康、拓扑替康),说明这类药物没有靶向递送加持已经有足够治疗窗口、这显然增加了ADC的成药机会。但除了安全窗口这个因素外喜树碱衍生物、尤其是DXd还有其它适合作为ADC毒素的性质。
喜树碱的发现起源于50年代在植物中寻找可的松衍生物的工作,后来才走上抗癌之路。喜树碱的关键药效团是E环的羟基内酯,这个内酯在生理pH值上下较小范围内就会可逆地快速开环关环、尤其是在体内或有人血清白蛋白(HSA)存在时。酸性条件(pH<7)催化关环、碱性条件(pH>7)催化开环,只要稍微偏离中性的pH7这些过程就可以在几分钟到几小时完成。开环的羟基羧酸比关环的内酯活性至少低10倍,但这两种存在形式都是有TOP1抑制活性的。羟基酸过膜性显然要比内酯差很多,所以杀伤活性的差别也可能是过膜性区别造成的。
伊利替康、拓扑替康这样的小分子药物打到静脉里因为血液pH>7所以很快就被水解成羟基酸,不仅活性丧失九成、而且羧酸与HAS结合率很高也很容易被肾脏排泄出人体,所以表观活性大幅度下降,这也是喜树碱没能进入早期ADC设计者法眼的原因之一。而ADC则是在细胞内的溶酶体里释放毒素,溶酶体是个酸性环境,所以无论是内酯还是羟基酸到了溶酶体统统都变成活性内酯了, Seagen的科学家证实喜树碱作为ADC毒素其内酯的稳定性与杀伤力无关。
这个释放机制一是提高了肿瘤细胞内活性内酯药物浓度,二是降低了肿瘤外的活性药物浓度。在进入溶酶体前喜树碱毒素即使内酯在循环系统被水解也因为连在抗体上无法被HAS或肾脏清除,而毒素在肿瘤释放后回到循环系统则会快速水解(碱性平衡态内酯浓度低于羟基酸盐),毒性降低、清除率提高。这几乎就是为ADC前药释放机制量身设计的软药型毒素,作为小分子药物的缺点改行作为ADC毒素因为起跑线环境变了也都变成优点了。另一类在内酯和羟基酸之间游荡的著名药物是他汀类降脂药,有的他汀是以内酯形式给药、有的则是以羟基酸盐形式给药。
溶酶体虽然是酸性,但是TOP1的工作场所细胞核和线粒体却是碱性环境,所以到了真正战场喜树碱毒素的存在形式仍是存疑的。因为开关环是个可逆过程所以搞清楚活性物质在TOP1活性腔中的存在形式并不容易,这两种形式不仅都有TOP1抑制活性、而且都有与TOP1形成复合物的晶体结构数据。羟基酸与内酯的极性和离子化完全不同,作为关键药效团活性相差只有10倍很不正常。或者TOP1这个酶就是为两种形式都有活性演化而来,或者是两种形式之间转化实在太快、失活代谢产物很难与活性药物区分开。
DXd虽然与依喜替康只有一个羟乙酰基的区别、而且这只是为了与GGFG偶联引入的一个adaptor,但是DXd似乎与上市喜树碱药物如SN38有一些重要差别。比如同样靶向Trop2,Data-DXd口腔炎发生率达到60%,但Trodelvy则发生率不到20%。T-DXd、Dato-DXd、HER3-DXd的间质性肺炎也都比Trodelvy发病率高。疗效方面T-DXd在HER2阴性乳腺癌和中枢转移乳腺癌的疗效也令人怀疑DXd是否有不同于主流喜树碱衍生物的药理学,因为结构高度类似的依喜替康即使做成释放非常平缓的高分子前药(DE310)也没有治疗窗口。当然现在也有依喜替康ADC进入临床、是否能成药我们拭目以待。
美中药源原创文章,转载注明出处并添加超链接,商业用途需经书面授权。★更多深度解析访问《美中药源》~
★ 请关注《美中药源》微信公众号 ★


:何为生物类似药?")


:生物类似药 VS. 化学仿制药")



- 路人丙: 新药发现的低悬果实
- Pipi_WHU: 新药发现的低悬果实
- 路人丙: Galapagos放弃IPF资产、吉利德空手而归
- Tan, Chengfang: Galapagos放弃IPF资产、吉利德空手而归
- 配体效率遭到质疑 | 美中药源: 你笑我无知类药性本不存在?
- Songdanqing: 你笑我无知类药性本不存在?
- 路人丙: 中国新药的新时代
- 康, 荣明: 中国新药的新时代
合作伙伴









 微信号:美中药源
微信号:美中药源