
基于片段药物设计(FBDD)简史
前天FDA批准了首个蛋白-蛋白相互抑制剂Venetoclax,这也是继2011年批准首个通过基于片段药物设计(FBDD)技术设计的药物—BRAF抑制剂Vemurafenib之后第二个成功利用FBDD设计的上市药物。今天我就简单讲讲FBDD技术。鉴于很多读者并非化学专业,所以我尽量通俗一点。
首先介绍几个基本概念。配体和蛋白的结合能力通常用抑制常数来衡量,而这个抑制常数和结合自由能成指数正相关。自由能量和能量的区别好比你的钱和所有钱的关系,自由能像你自己财产一样可以用来做事。所有化学过程都受热力学第二定律控制,这个定律指出在封闭体系任何自发过程自由能必然降低,翻译成大白话就是如果你不是富二代(没有能量输入,即封闭体系)任何你享受的事情必然导致你财产减少。化合物活性的提高本质上是化合物与靶点结合后体系自由能降低幅度的提高。
结合自由能和很多因素有关,但简单地说分子越大、结合位点越多,结合能力越强。大概相当于消费方式越多消费者越高兴。90年代多个研究显示如果选择恰当,在一定范围内每增加一个重原子结合自由能最多可以增加1.5Kcal/mol,大概相当于提高11倍结活性。注意这不意味着随便在分子上加任何重原子都能提高11倍活性,而是根据经验最多可以增加这么多活性。这个逻辑框架也是现在FBDD优化必须跟踪的一个指标、配体效率(LE)的基础。
早期的新药开发没有靶点,也自然谈不上结合能。现在公认的首个理性设计药物是H2受体拮抗剂甲氰咪胍,当时的先导物是内源性配体组胺。后来ACE抑制剂的成功使这种优化化合物和单个靶点结合强度的新模式流行起来。ACE的先导物来自巴西蛇毒里分出的一个多肽。无论组胺(本身是个激动剂)还是多肽优化成最后药物都非常不容易,而且对很多靶点来说即使这样难于优化的先导物也没有。
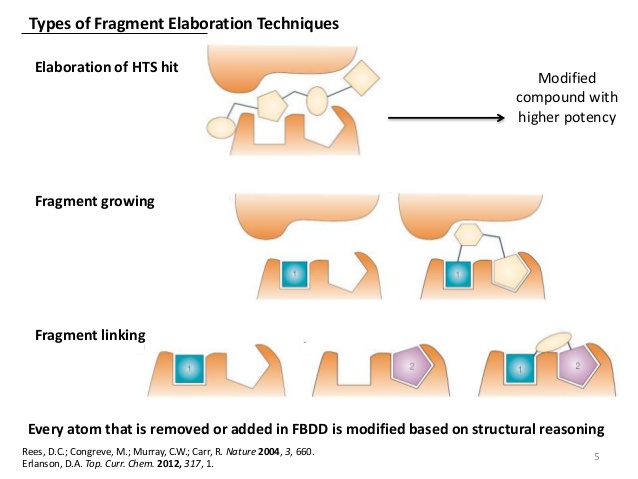
所以90年代中期有人想到FBDD这个思路。想法是虽然活性很好的先导物不好找,但结合能力弱的先导物还是容易找。如果能把两个弱结合片段链接在一起,结合能则可以加到一起。因为活性和结合能是指数正相关,所以活性可以指数提高。比如一个1 uM的配体如果和一个很弱的1 mM配体链接起来可能产生1 nM的高强度配体。最早的所谓片段链接技术就是通过同位素标记的蛋白筛选能和这个蛋白不同位点结合的片段,然后用化学手段不两个片段结合在一起成为高活性配体。
2001年GSK的Michael Hann用一个非常简单的模型指出理论上越小的片段筛选成功率越高,所以FBDD更高效。但这个模型假设配体和蛋白的作用非正即负,事实上并非所有系统都这样。有些靶点适合FBDD,而有些靶点不适合。FBDD永远不可能发现吗啡这样的复杂配体。2006年Shoichet把一个高活性配体一段段肢解到片段,结果每剪短一部分后的分子都和原来分子与同一蛋白的结合方式不同。所以如果从最小片段开始用FBDD技术将很难发现开始的高活性配体。
如果传统的配体/靶点结合算是锁头和钥匙的关系,那么FBDD好比用腻子密封窗子。虽然很长,但是每段都很简单,基本和搭积木类似。如上面所讲,FBDD很难把片段做成复杂的钥匙。虽然上腻子相对简单,但还是需要一些有关窗子形状的信息。即使是PPI这种表面没有什么结构特征的结合腔,一般FBDD也必须有晶体结构或核磁结构的支持,以减少需要合成的化合物。否则优化可能性太多,需要合成的化合物太多。有些人宣称没有蛋白结构也成功使用FBDD发现高活性配体,这种情况多是从文献借鉴了大部分类似分子的结构特征。把甲基接到吗啡上得到的活性配体不能算是从甲烷开始的FBDD设计。
在FBDD开始的时候Lipinski也提出了著名的五规则。FBDD最大的问题是起点化合物活性很低,如果不严格控制将需要很大的分子才能达到需要的活性。而分子太大则吸收溶解都不好,对于细胞内靶点还有过膜问题。所以FBDD必须监控配体效率以保证LE不会随着活性提高而下降。但因为适于FBDD的靶点通常需要超出传统药物分子大小的配体,所以FBDD发现的上市药物并不多,这个模式也不可能取代HTS用于所有的靶点。
推荐阅读
美中药源原创文章,转载注明出处并添加超链接,商业用途需经书面授权。★更多深度解析访问《美中药源》~
★ 请关注《美中药源》微信公众号 ★


:何为生物类似药?")


:生物类似药 VS. 化学仿制药")



- 路人丙: 新药发现的低悬果实
- Pipi_WHU: 新药发现的低悬果实
- 路人丙: Galapagos放弃IPF资产、吉利德空手而归
- Tan, Chengfang: Galapagos放弃IPF资产、吉利德空手而归
- 配体效率遭到质疑 | 美中药源: 你笑我无知类药性本不存在?
- Songdanqing: 你笑我无知类药性本不存在?
- 路人丙: 中国新药的新时代
- 康, 荣明: 中国新药的新时代
合作伙伴









 微信号:美中药源
微信号:美中药源